写在前面
同之前一样,重分析需要所以自己找了各路代码借鉴学习,详情请参考 R语言绘制带聚类树的堆叠柱形图 , 实操效果如下:

步骤
表格预处理
- 选取不同样本属水平的物种丰度图(绝对和相对水平都可以,相对画出来是齐的,绝对的bar是不齐的)。Top10丰度外的类群合并为Others。这里注意,用本篇代码时,表格选择csv/txt等格式文件一定要是制表符分隔的,不然会报错重复名或者没有多出的行
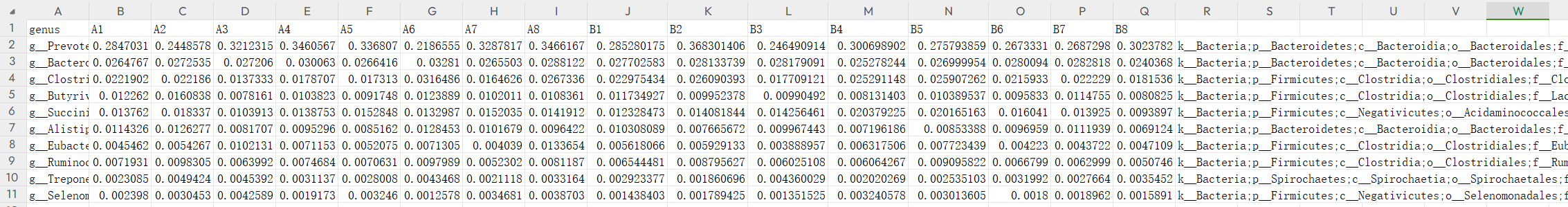
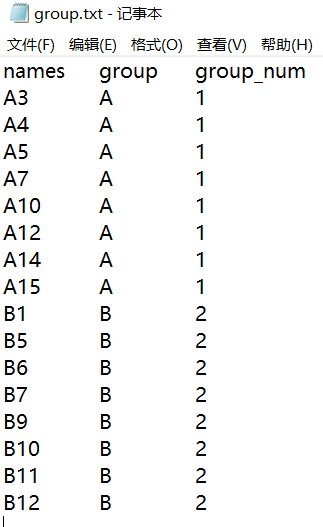
- 分组信息
代码演示
# 装包
install.packages("vegan")
#层次聚类
#读取 OTU 丰度表,csv/txt等文件一定要是制表符分隔的,不然会报错重复名或者没有多出的行
dat <- read.delim('Unigenes.absolute.g.txt', row.names = 1, sep = '\t', head = TRUE, check.names = FALSE)
#计算样本间距离,以群落分析中常用的 Bray-curtis 距离为例
dis_bray <- vegan::vegdist(t(dat), method = 'bray')
#层次聚类,以 UPGMA 为例
tree <- hclust(dis_bray, method = 'average')
tree
plot(tree)
##聚类树绘制
#样本分组颜色、名称等
group <- read.delim('group.txt', row.names = 1, sep = '\t', head = TRUE, check.names = FALSE, stringsAsFactors = FALSE)
grp <- group[2]
group_col <- c('red', 'blue')
names(group_col) <- c('1', '2')
group_name <- c('RA', 'CON')
#样本分组标签
layout(t(c(1, 2, 2, 2, 3)))
par(mar = c(5, 2, 5, 0))
plot(0, type = 'n', xaxt = 'n', yaxt = 'n', frame.plot = FALSE, xlab = '', ylab = '',
xlim = c(-max(tree$height), 0), ylim = c(0, length(tree$order)))
legend('topleft', legend = group_name, pch = 15, col = group_col, bty = 'n', cex = 1)
#聚类树绘制,按分组给分支上色
treeline <- function(pos1, pos2, height, col1, col2) {
meanpos = (pos1[1] + pos2[1]) / 2
segments(y0 = pos1[1] - 0.4, x0 = -pos1[2], y1 = pos1[1] - 0.4, x1 = -height, col = col1,lwd = 2)
segments(y0 = pos1[1] - 0.4, x0 = -height, y1 = meanpos - 0.4, x1 = -height, col = col1,lwd = 2)
segments(y0 = meanpos - 0.4, x0 = -height, y1 = pos2[1] - 0.4, x1 = -height, col = col2,lwd = 2)
segments(y0 = pos2[1] - 0.4, x0 = -height, y1 = pos2[1] - 0.4, x1 = -pos2[2], col = col2,lwd = 2)
}
meanpos = matrix(rep(0, 2 * length(tree$order)), ncol = 2)
meancol = rep(0, length(tree$order))
for (step in 1:nrow(tree$merge)) {
if(tree$merge[step, 1] < 0){
pos1 <- c(which(tree$order == -tree$merge[step, 1]), 0)
col1 <- group_col[as.character(grp[tree$labels[-tree$merge[step, 1]],1])]
} else {
pos1 <- meanpos[tree$merge[step, 1], ]
col1 <- meancol[tree$merge[step, 1]]
}
if (tree$merge[step, 2] < 0) {
pos2 <- c(which(tree$order == -tree$merge[step, 2]), 0)
col2 <- group_col[as.character(grp[tree$labels[-tree$merge[step, 2]],1])]
} else {
pos2 <- meanpos[tree$merge[step, 2], ]
col2 <- meancol[tree$merge[step, 2]]
}
height <- tree$height[step]
treeline(pos1, pos2, height, col1, col2)
meanpos[step, ] <- c((pos1[1] + pos2[1]) / 2, height)
if (col1 == col2) meancol[step] <- col1 else meancol[step] <- 'grey'
}
##堆叠柱形图
#样本顺序调整为和聚类树中的顺序一致
dat <- dat[ ,tree$order]
#物种颜色设置
genus_color <- c('#8DD3C7', '#FFFFB3', '#00CDCD', '#FB8072', '#80B1D3', '#FDB462', '#B3DE69', '#FCCDE5', '#BC80BD', '#CCEBC5', 'gray')
names(genus_color) <- rownames(dat)
#堆叠柱形图
#数字向量,格式为c(bottom, left, top, right),给出各个方向绘图区边缘空白的大小
par(mar = c(5, 3, 5, 0))
relative <- read.delim('new_Unigenes.relative.g.txt', row.names = 1, sep = '\t', head = TRUE, check.names = FALSE)
bar <- barplot(as.matrix(relative), col = genus_color, space = 0.4, width = 0.7, cex.axis = 1, horiz = TRUE, cex.lab = 1.2,
xlab = 'Relative Abundance', yaxt = 'n', las = 1, ylim = c(0, ncol(dat)), family = 'mono')
mtext('Top 10 genus', side = 3, line = 1, cex = 1)
text(x = -0.05, y = bar, labels = colnames(dat), col = group_col[group[tree$order, 2]], xpd = TRUE)
#柱形图图例
par(mar = c(5, 1, 5, 0))
plot(0, type = 'n', xaxt = 'n', yaxt = 'n', bty = 'n', xlab = '', ylab = '')
legend('left', pch = 15, col = genus_color, legend = names(genus_color), bty = 'n', cex = 1)